The Probability Geometry of Preclinical Bets: Why Your Expected Value Model Is Probably Wrong (And Why You Should Build One Anyway)
Quick Links: Knowledge Base, Podcast, and Social
Knowledge Base — search and filter every article and podcast episode by topic, section, and keyword: kb.onhealthcare.tech
Listen to the Podcast — every article is also available as an audio episode. Free subscribers get the public episodes; paid subscribers get the full archive including subscriber-only episodes. Listen on Apple Podcasts, Spotify, or browse all episodes on the Substack Podcast page.
For paid subscribers — your subscription unlocks the entire research archive (538+ deep-dives), every paid podcast episode, and full search inside the Knowledge Base. To listen to paid episodes in Apple or Spotify, link your Substack subscription via the show settings on those platforms (instructions inside the Substack app under Subscriptions → Podcast).
For free subscribers — free posts and free podcast episodes are always public on Apple/Spotify and Substack. Upgrade any time at onhealthcare.tech/subscribe to access the paid archive and paid episodes.
Follow on Social — X · YouTube · TikTok · Instagram
Table of Contents
Introduction: The Seductive Illusion of Precision
The Fundamental Architecture of Preclinical Expected Value
Why Traditional Models Systematically Fail
The Hidden Variables: What Actually Drives Outcomes
Temporal Discounting and the Patience Premium
Portfolio Construction in a Power Law World
The Sociology of Scientific Risk
Practical Implementation: Building Models That Actually Work
Conclusion: Embracing Productive Uncertainty
Abstract
This essay examines the theoretical foundations and practical challenges of modeling expected value in preclinical biotech investments, with particular emphasis on the systematic biases that plague traditional approaches. Key themes include:
- The mathematical structure of preclinical expected value models and their sensitivity to input assumptions
- Empirical data on success rates, value creation, and timeline distributions across therapeutic modalities
- The role of hidden variables, information asymmetry, and scientific sociology in outcome determination
- Portfolio construction strategies that account for power law distributions and correlation structures
- Practical frameworks for building useful models despite irreducible uncertainty
Introduction: The Seductive Illusion of Precision
There exists a particular species of spreadsheet that haunts the conference rooms of venture capital firms, circulates through the inboxes of limited partners, and justifies the deployment of hundreds of millions of dollars into molecules that have never touched human tissue. This spreadsheet purports to calculate expected value for preclinical biotech investments with impressive specificity, often extending to decimal points that would make a quantum physicist blush. The model multiplies probability of technical success by probability of regulatory success by probability of commercial success, applies a discount rate, subtracts invested capital, and produces a number that feels reassuringly concrete. The problem, of course, is that this number is almost certainly wrong, and more dangerously, wrong in ways that are highly predictable and systematically biased.
The challenge of modeling expected value in preclinical biotech combines the worst aspects of venture capital, pharmaceutical development, and predictive modeling. You are attempting to forecast outcomes that depend on scientific discoveries not yet made, regulatory decisions not yet contemplated, and competitive dynamics not yet emerged, all while operating with incomplete information about the fundamental biology, chemistry, and market structure. The modal preclinical asset will fail, and fail expensively. The handful that succeed will do so through pathways that were largely unforeseeable at the time of initial investment. And yet, despite this fundamental uncertainty, capital must be allocated, decisions must be made, and some framework for thinking about relative value must be constructed.
This essay argues that the solution is not to abandon expected value modeling but rather to understand its limitations, acknowledge its biases, and build models that are useful rather than precise. The goal is not to predict the future with accuracy but to think clearly about uncertainty, to identify the variables that actually matter, and to construct portfolios that are robust to our inevitable misconceptions about how biology and business intersect.
The Fundamental Architecture of Preclinical Expected Value
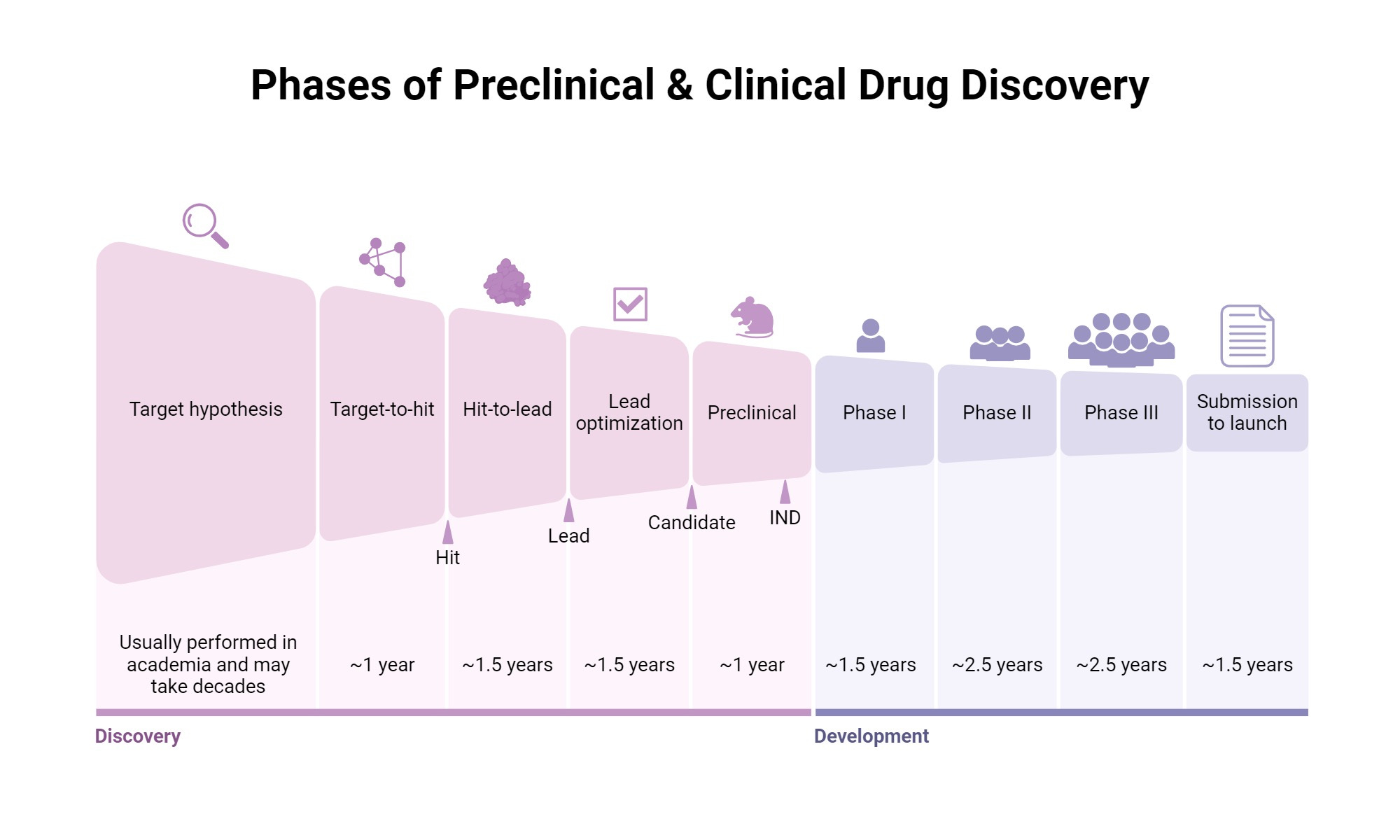
At its core, expected value modeling for preclinical biotech follows a straightforward multiplicative structure. You begin with some estimate of the probability that the molecule will successfully navigate each stage of development: preclinical safety and efficacy, Phase One dose escalation and safety, Phase Two proof of concept, Phase Three pivotal trials, regulatory approval, and commercial launch. You then estimate the potential value if the asset reaches the market, typically through some combination of peak sales projections, market share assumptions, and comparable transaction multiples. Finally, you discount this terminal value back to the present using an appropriate discount rate, subtract the capital required to reach that outcome, and arrive at a net present value.
The mathematics are simple enough that they can be executed on a napkin. A thirty percent probability of preclinical success, multiplied by sixty percent probability of Phase One success, multiplied by thirty percent probability of Phase Two success, multiplied by seventy percent probability of Phase Three success, multiplied by ninety percent probability of regulatory approval, yields an overall probability of approximately three point four percent. If the terminal value is two billion dollars and the discount rate is fifteen percent applied over twelve years, the present value of success is approximately three hundred fifty million dollars. Multiply by the three point four percent probability and you get roughly twelve million dollars in expected value. Subtract the hundred fifty million dollars you will spend getting there and you have massively negative expected value, which is why you should not fund this asset.
Except, of course, that every single number in that calculation is suspect. The probability estimates are derived from historical databases that aggregate radically different therapeutic modalities, indications, and competitive contexts. The terminal value calculation assumes a static market, unchanged competitive dynamics, and perfect execution on commercialization. The discount rate is borrowed from corporate finance theory that was developed for mature industrial businesses, not venture-scale biotechnology. And the entire framework assumes that these probabilities are independent, that success at one stage does not meaningfully update your beliefs about success at subsequent stages, which is manifestly false for any asset where mechanism of action and target biology matter.
Why Traditional Models Systematically Fail
The systematic failures of traditional expected value models in preclinical biotech stem from three fundamental sources: information compression, selection bias, and temporal myopia. Each of these biases operates in predictable ways, and understanding them is essential to building better frameworks.
Information compression occurs when rich, multidimensional data about an asset is reduced to a single probability estimate. Consider what is actually known about a preclinical oncology asset targeting a novel kinase in non-small cell lung cancer. You have preclinical efficacy data in multiple cell lines and mouse models. You have pharmacokinetic and pharmacodynamic data describing drug exposure and target engagement. You have competitive intelligence about other drugs in the same pathway. You have scientific literature about the role of this kinase in cancer progression and resistance mechanisms. You have regulatory precedent for this indication and this mechanism class. Compressing all of this information into a single number labeled “probability of Phase Two success equals thirty percent” discards almost everything that might actually distinguish this asset from the base rate.
The better approach recognizes that what matters is not the base rate but rather how this specific asset differs from the base rate, and in which direction. A novel mechanism with sparse preclinical validation but extraordinary early efficacy signals should be modeled differently than a fast-follower asset in a well-validated pathway with competitive but unexceptional data. The probability distribution for the former is wider and more skewed, the correlation between stages is lower, and the value if successful is potentially much higher. Traditional models that rely on point estimates for stage-wise probabilities cannot capture these distinctions.
Selection bias represents perhaps the most pernicious systematic error in preclinical expected value models. The historical databases from which probability estimates are derived are drawn from assets that were selected for clinical development by prior generations of investors and management teams. These assets were not randomly selected from the universe of possible drug candidates but rather were chosen precisely because they appeared to have better than average probability of success. This means that the base rates you are using for your model are already conditioned on positive selection, and applying them naively to your asset overstates its probability of success unless you have strong reasons to believe your selection process is more rigorous than the historical average.
The magnitude of this bias is difficult to quantify but almost certainly substantial. If historical Phase Two success rates for oncology are approximately thirty percent, but these assets were selected from a broader pool where the true success rate would have been ten percent without selection, then using thirty percent as your base rate implies that your diligence process adds no value relative to historical norms. In reality, some portion of that thirty percent success rate is attributable to selection, some portion is attributable to genuine improvements in scientific understanding and technological capability over time, and only the residual is available as a reasonable prior for your specific asset.
Temporal myopia manifests in the systematic underweighting of long-duration risks and opportunities. Standard expected value models apply a constant discount rate over the entire development timeline, which implicitly assumes that a dollar of value realized in twelve years is worth precisely the same as a dollar of value realized in twelve years regardless of the path taken to get there. This is clearly wrong. An asset that reaches Phase Three and fails is vastly more informative and strategically valuable than an asset that fails in Phase One, even if both ultimately return zero. The option value of information, the ability to make course corrections, and the potential for platform learning are all ignored in simple discounted cash flow frameworks.
Moreover, the discount rates typically applied in venture-scale biotech, ranging from fifteen to forty percent depending on stage and risk profile, are borrowed from frameworks designed for mature businesses with relatively stable cash flows. These discount rates are meant to capture the time value of money, the risk of total loss, and the opportunity cost of alternative investments. But in preclinical biotech, the dominant risk is not that the world will change but that your scientific hypothesis will prove false. This is a different kind of risk than market risk, and it is not clear that it should be modeled using the same mathematical machinery.
The Hidden Variables: What Actually Drives Outcomes
If traditional expected value models systematically fail to predict outcomes, what variables actually matter? Empirical analysis of preclinical biotech investments suggests that a relatively small number of factors explain a disproportionate share of variance in outcomes, and these factors are often poorly captured in standard models.
Target validation quality stands out as perhaps the most important predictor of clinical success that is routinely underweighted in expected value calculations. Assets targeting mechanisms with strong human genetic evidence of causality, where loss of function mutations produce the desired phenotype and gain of function mutations produce the disease phenotype, have dramatically higher success rates than assets based on purely preclinical or correlative evidence. A 2019 analysis published in Nature Genetics found that drug development programs supported by genetic evidence were approximately twice as likely to reach regulatory approval compared to programs without such evidence. Yet standard models typically treat all Phase One assets in a given indication as having similar probability of success, ignoring this fundamental difference in biological validation.
The quality of target validation varies enormously across therapeutic areas. In oncology, the revolution in genomic profiling has enabled target selection based on driver mutations and synthetic lethality relationships, dramatically improving the informativeness of preclinical models. In neuroscience, by contrast, the gap between rodent models and human disease remains vast, and target validation is correspondingly weaker. Applying the same base rate assumptions across these contexts ignores information that has substantial predictive power.
Team quality and organizational learning represent another cluster of variables that are difficult to quantify but enormously important in practice. Preclinical development is not a deterministic process of moving molecules through predefined checkpoints but rather an iterative process of hypothesis generation, experimental design, interpretation, and course correction. Teams that have successfully navigated this process before, that understand which experiments are truly informative versus merely impressive, and that maintain appropriate skepticism about their own data, produce systematically better outcomes than teams executing their first program.
The challenge in modeling team quality is that it requires moving beyond credentials and pedigree to assess actual capability at the specific tasks of preclinical drug development. A founding team with impressive academic publications and prior exits may or may not have the specific skills required to design a toxicology study, interpret off-target binding data, or navigate a pre-IND meeting with the FDA. Conversely, teams with less prominent backgrounds but deep operational experience in drug development may be substantially de-risked relative to what standard models would suggest.
Competitive dynamics and market timing introduce another layer of complexity that is poorly captured in static expected value models. The value of a preclinical asset depends not only on its intrinsic probability of technical success but also on the landscape of competing programs, the timing of their clinical readouts, and the evolution of standard of care. An asset that is first-in-class with a novel mechanism has a radically different risk-reward profile than a fifth-in-class asset in a crowded pathway, even if the underlying biology is similar.
The temporal dimension of competitive dynamics is particularly important and frequently ignored. Clinical development timelines are long and uncertain, which means that the competitive landscape at the time of your potential launch is fundamentally unknowable at the time of preclinical investment. An asset that begins development in a sparse competitive landscape may reach Phase Three only to discover that multiple competing programs have reported positive data, collapsed peak sales projections, and compressed valuations. Conversely, assets that initially appear to face substantial competition may benefit from competitive failures, emerging resistance mechanisms, or market expansion.
Temporal Discounting and the Patience Premium
The application of discount rates to preclinical expected value calculations deserves particular scrutiny because it embeds assumptions about time preference, risk, and opportunity cost that are often inappropriate for venture-scale biotechnology. Traditional corporate finance theory suggests that discount rates should reflect the systematic risk of an asset, typically measured by beta in the capital asset pricing model framework. But preclinical biotech returns are driven primarily by idiosyncratic scientific risk rather than market risk, which suggests that standard CAPM-derived discount rates are misspecified.
An alternative framework recognizes that the primary cost of capital in preclinical biotech is not the opportunity cost of alternative financial investments but rather the opportunity cost of alternative deployment of scarce scientific and operational resources. In this view, the appropriate discount rate should reflect the rate at which alternative preclinical opportunities are becoming available, the rate at which scientific knowledge is accumulating, and the degree to which delayed entry into clinical development creates strategic disadvantage.
This perspective suggests that discount rates for preclinical biotech should potentially be lower than those typically applied, particularly for assets in areas where scientific understanding is improving rapidly and where being second-to-market is dramatically worse than being first-to-market. The patience premium, the additional expected value available to investors willing to fund longer-duration development programs, may be substantial in therapeutic areas where most capital is focused on late-stage, near-term opportunities.
Consider the difference between a gene therapy program requiring five years of preclinical development to optimize manufacturing and delivery versus a small molecule program that could enter Phase One within eighteen months. Standard expected value models would heavily penalize the gene therapy program for its longer timeline, applying an additional three and a half years of discounting that might reduce present value by thirty to forty percent. But if the gene therapy has the potential to be curative while the small molecule is merely palliative, if the competitive landscape for gene therapy is less crowded, and if manufacturing optimization creates durable competitive advantages, then the longer timeline might actually be a feature rather than a bug.
Portfolio Construction in a Power Law World
The distribution of returns in preclinical biotech follows a power law, where a small number of extreme outcomes account for the majority of total value creation. This has profound implications for portfolio construction that are poorly captured in expected value models that focus on individual assets in isolation. In a power law world, the primary objective is not to avoid losses, which are inevitable and frequent, but rather to ensure exposure to the tail outcomes that drive portfolio-level returns.
Data from biotechnology venture capital suggests that the top ten percent of investments generate approximately eighty to ninety percent of total returns, and the top one percent of investments may generate thirty to forty percent of total returns. This distribution is substantially more skewed than in software venture capital and dramatically more skewed than in traditional private equity. The implication is that portfolio construction should be oriented around maximizing the probability of capturing at least one or two extreme positive outcomes rather than minimizing the frequency of losses.
This suggests several non-obvious portfolio construction principles. First, portfolio size should be large enough to provide reasonable statistical power for capturing tail outcomes, which probably means fifteen to thirty preclinical investments for a dedicated fund. Second, position sizing should be relatively uniform rather than concentrated, because the ex-ante difficulty of identifying which assets will produce extreme outcomes argues against aggressive concentration. Third, reserves should be managed to ensure the ability to fund winners through to meaningful value inflection points, even if this requires abandoning some investments that are merely performing adequately.
The correlation structure of preclinical biotech portfolios deserves particular attention. Assets that share common mechanisms, targets, or therapeutic areas have positive correlation in their outcome distributions, which reduces portfolio-level diversification. Assets that depend on shared scientific hypotheses, such as the druggability of a particular protein family or the validity of a particular disease model, have even higher correlation. Building portfolios that are diversified across truly independent scientific hypotheses is substantially more difficult than simply diversifying across indications or modalities.
Conversely, some forms of correlation are desirable. Assets that share common operational capabilities, such as manufacturing platforms, delivery technologies, or regulatory strategies, may have positive correlation but also generate learning spillovers that improve outcomes across the portfolio. The optimal portfolio balances hypothesis diversification with operational leverage, ensuring that both positive and negative information from early investments can be productively deployed to improve subsequent investment decisions.
The Sociology of Scientific Risk
One of the most underappreciated dimensions of preclinical biotech risk is the role of scientific culture, incentive structures, and social dynamics in determining outcomes. Preclinical development occurs within a particular sociological context, where scientists have career incentives that may or may not align with accurate risk assessment, where organizational structures shape information flow and decision-making, and where broader scientific communities establish norms about what constitutes sufficient evidence for advancing molecules into clinical development.
Academic science, from which most preclinical biotechnology originates, operates under incentive structures that reward novel findings, surprising results, and mechanistic insights. These incentives produce enormous value in terms of expanding the frontiers of knowledge but create systematic biases when applied to drug development. Academic publications are selected for statistical significance and novelty, which means that the literature systematically overrepresents positive results and underrepresents null results. Preclinical models that work well enough to generate publications may not predict clinical outcomes with anything approaching the fidelity suggested by published data.
The translation of academic science into biotechnology companies introduces additional layers of complexity. Founding scientists often maintain dual affiliations with academic institutions and companies, creating potential conflicts between the norms of academic publishing and the requirements of competitive drug development. Early employees are frequently drawn from academic laboratories where the cultural emphasis is on intellectual creativity rather than operational rigor. Management teams must navigate the tension between maintaining scientific enthusiasm and imposing the discipline required for successful drug development.
These sociological dynamics manifest in predictable failure modes. Companies may advance molecules into clinical development based on incomplete preclinical validation because the founding scientists are intellectually committed to a particular hypothesis. Key experiments that would genuinely de-risk the program may be postponed because they are scientifically uninteresting or because negative results would undermine the company’s narrative. Decision-making may be captured by the most scientifically senior or charismatic individuals rather than being driven by systematic evaluation of evidence.
The better biotechnology organizations develop cultures that explicitly counteract these biases. They create incentives for rigorous rather than creative science, reward the identification of problems as much as the generation of solutions, and maintain clear separation between the scientific validation of hypotheses and the business development of assets. They invest heavily in experiments that could disprove their core hypotheses rather than focusing exclusively on experiments that confirm and extend their initial findings. And they cultivate comfort with ambiguity and uncertainty rather than premature confidence in scientific narratives.
Practical Implementation: Building Models That Actually Work
Given the limitations of traditional expected value models, what does a practically useful framework for preclinical biotech investment look like? The answer is not to abandon quantitative modeling but rather to build models that acknowledge uncertainty, incorporate rich information about what actually drives outcomes, and support decision-making under conditions of irreducible ambiguity.
The foundation of a useful model is explicit representation of uncertainty through probability distributions rather than point estimates. Instead of modeling Phase Two success probability as thirty percent, model it as a beta distribution with appropriate parameters that capture both the expected value and the uncertainty around that expectation. This enables Monte Carlo simulation that produces a distribution of outcomes rather than a single expected value, making clear the range of possibilities and the sensitivity to input assumptions.
Incorporating conditional probabilities and Bayesian updating represents another significant improvement over static models. The probability of Phase Three success conditional on Phase Two success is substantially higher than the unconditional probability of Phase Three success, because positive Phase Two data updates your beliefs about the underlying biology, the quality of the clinical endpoints, and the adequacy of the dose. Modeling these conditional relationships explicitly, and updating them as new information becomes available, produces expected value estimates that are much more responsive to the actual evolution of the asset.
Multi-factor models that decompose success probability into underlying components provide richer insight than aggregate stage-wise probabilities. Rather than estimating Phase Two success as a single number, decompose it into the probability that the mechanism is correct, the probability that the dose is adequate, the probability that the endpoints are informative, the probability that the patient population is well-selected, and so forth. This decomposition serves two purposes: it forces explicit reasoning about what would need to be true for the asset to succeed, and it enables more targeted updating as new information becomes available.
Scenario analysis and stress testing should be routine rather than exceptional. Every expected value model should be accompanied by explicit consideration of alternative scenarios: what if the competitive landscape evolves differently, what if manufacturing costs are higher, what if the indication proves smaller than expected. These scenarios should not be treated as mere sensitivity analysis but rather as explicit hypotheses about alternative futures, each with associated probabilities and implications for decision-making.
Real options analysis provides a framework for valuing the optionality and flexibility inherent in preclinical development. The ability to expand into additional indications, modify the molecule based on early clinical data, or pivot to alternative mechanisms based on competitive developments all create value that is not captured in simple discounted cash flow models. Explicitly valuing these options, even approximately, provides a more complete picture of expected value.
Conclusion: Embracing Productive Uncertainty
The central argument of this essay is that expected value modeling in preclinical biotech should be understood not as a forecasting exercise but as a framework for thinking clearly about uncertainty. The models we build are wrong, they will always be wrong, and they will be wrong in ways that are systematic and predictable. But this does not make them useless. A model that makes explicit our assumptions, quantifies our uncertainty, and enables systematic comparison across opportunities is valuable even if its point estimates prove inaccurate.
The goal is to build models that are useful rather than precise, models that capture the variables that actually drive outcomes, and models that support better decision-making under conditions of irreducible uncertainty. This requires acknowledging the limitations of historical base rates, incorporating rich information about target validation and team quality, recognizing the power law distribution of outcomes, and understanding the sociological context in which scientific risk is actually realized.
It also requires cultivating an appropriate relationship with quantitative modeling more broadly. Models should inform judgment rather than replace it, should be updated as new information becomes available rather than treated as static, and should be understood as provisional frameworks rather than objective truths. The investors and entrepreneurs who do this well combine quantitative rigor with qualitative judgment, using models to sharpen their thinking while remaining alert to what the models miss.
In the end, preclinical biotech investment is an exercise in making decisions with incomplete information about systems that are too complex to fully understand, in pursuit of outcomes that are too rare to predict with statistical confidence. Expected value modeling provides a language for discussing these decisions, a framework for thinking about risk and return, and a basis for learning from both successes and failures. That is enough. The alternative, making decisions based on intuition alone or deploying capital without any systematic framework for evaluation, is strictly worse. Build the model, understand its limitations, update it as you learn, and make decisions that are robust to the ways in which the model will inevitably prove wrong. This is the art of preclinical biotech investment, and no spreadsheet will ever fully capture it.